DeepFeature
DeepFeature converts non-image samples into image-form and performs element selection via convolutional neural network (CNN). This approach builds an image by arranging elements (or genes) by finding similarity among them and then by mapping the non-image values on to these aligned pixel locations. This approach employs CNN for element or gene selection on non-image data. A real world application of DeepFeature to publicly available cancer data identified gene sets with significant overlap to several cancer-associated pathways suggesting the potential of this method to discover biomedically meaningful connections.
Reference
Sharma A, Lysenko A, Boroevich KA, Vans E, Tsunoda T, DeepFeature: feature selection in nonimage data using convolutional neural network, Briefings in Bioinformatics, 22(6), bbab297, 2021
Download and Install
-
Download Matlab package DeepFeature_pkg.tar.gz from the link above. Store it in your working directory. Gunzip and untar as follows:
>> gunzip DeepFeature_pkg.tar.gz >> tar -xvf DeepFeature_pkg.tar -
Follow the link: RNA-Seq Data to download the RNA-seq data (caution: data size is 1.5GB). The dataset is given in .mat file format of Matlab. Place the data file ‘dataset1.mat’ in the folder,
~/DeepFeature_pkg/Data/ -
Download and Install Squeezenet in Matlab, see details about Squeezenet from MathWorks link.
Run to check if the package is installed correctly
-
To check and run the package in a faster way,
-
Go to Line 20 of DeepInsight_train_norm_CAM.m file and replace ‘MaxObj’,50,…, to ‘MaxObj’,2,… (This will allow only 2 objective functions to run on GPU).
-
Go to Line 18 of DeepFeature.m and change
Parm.DesiredGenes = 8000. This will run the code in one loop otherwise next loop with continue.
-
-
Open the main file, DeepFeature.m and Run. A few messages will be displayed on the Command Window of Matlab, such as
Stage 2 Begins Starting Data preparation by DeepInsight Dataset: RNAseq NORM-2 tSNE with burneshut algorithm has been used Pixels: 227 x 227 Input Size 1 x Input Size 2: 227 x 227 Data Preparation ends Training model beginsNote that the below values might differ.
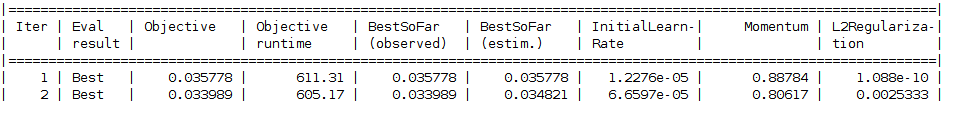
Objective function image will be shown for the Bayesian Optimization Technique
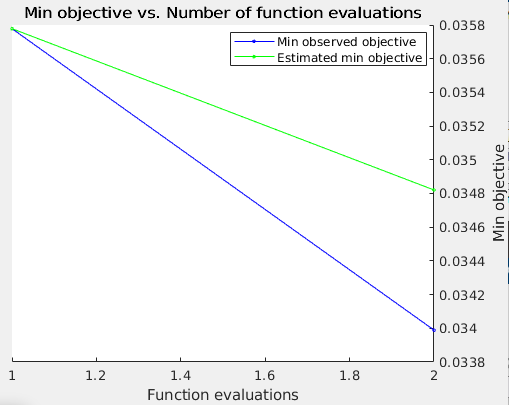
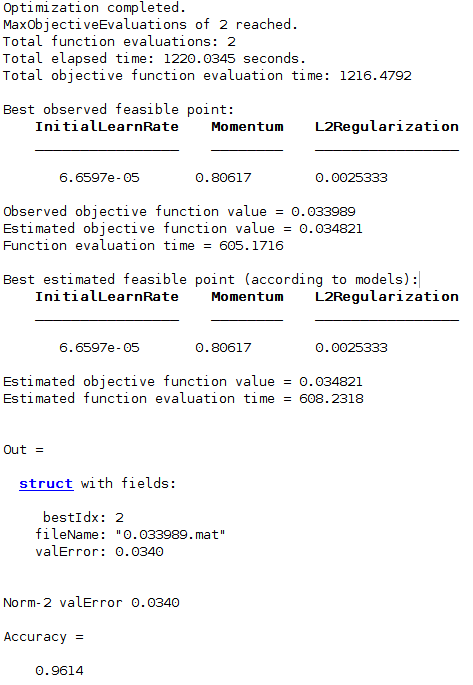
Stage: 2; Test Accuracy: 0.9614; ValErr: 0.0340; Momentum: 0.806168; L2Regularization: 0.00253334; InitLearnRate: 6.65972e-05 Training model ends Feature selection begins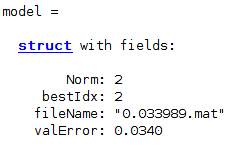
Starting parallel pool (parpool) using the ‘local’ profile . . . Connected to the parallel pool (number of workers: 20).Some images will be shown, e.g.:
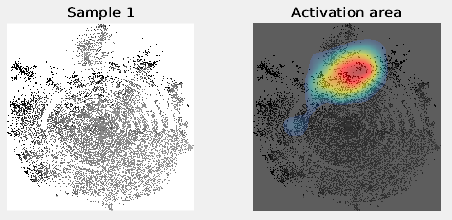


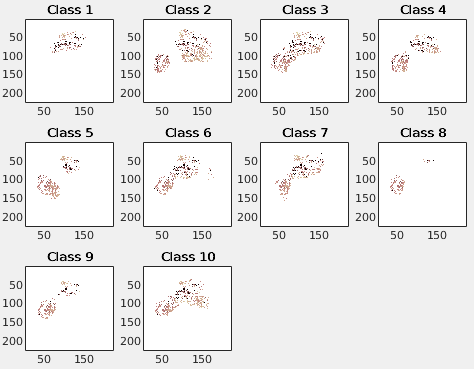
TIME = 770.6822 Saved … Files Saved … #Genes = 5539; #Genes_compress = 3613 Feature selection ends Stage 2 Ends
Note:
-
All the results will be stored in current stage folder
~/DeepFeature_pkg/Models/Run1/StageXwhere X is the current stage; i.e., ‘2’ here. -
Similarly, all the figures will be stored in a folder
~/DeepFeature_pkg/FIGS/Run1/StageXwhere X is the current stage; i.e., ‘2’ here. -
If the loop continues then the value of X will increment to 3, 4, …; i.e., repeating DeepFeature model to find a smaller subset of genes.
Description of files and folders
-
DeepFeature_pkg has 4 folders: Data, DeepResults, FIGS and Models. It has several .m files. However, the main file is ‘DeepFeature.m’. All the parameter settings can be done in this file.
-
DeepFeature.m has 3 main functions:
-
func_Prepare_Data: This function loads the data, splits the training data into the Train and Validation sets, normalizes all the 3 sets (including Test set), and converts non-image samples to image form using the Train set. The Test and Validation sets are not used to find pixel locations. Once the pixel locations are obtained, all the non-image samples are converted to image samples. The image datasets are stored as Out1.mat or Out2.mat. -
func_TrainModel: This function executes the convolution neural network (CNN) using SqueezeNet architecture. However, the user may change the net as required. It optimizes hyper-parameters using the Bayesian Optimization Technique. The nets are modified so that feature selection can be performed. It uses Train set and Validation set to tune and evaluate the model hyper-parameters.Note: To test the code more quickly (as discussed above), please replace the number of the maximum objective function to 1 or 2 in Line 20 of DeepInsight_train_norm_CAM.m file. By default it is fifty; i.e., ‘MaxObj’,50,…, change to ‘MaxObj’,2,… (for example).
The best evaluation is stored in DeepResults folder as .mat files, where the file name depicts the best validation error achieved. For example, file 0.035778.mat in DeepResults folder tells the hyper-parameters at validation error 0.035778. Also, the model file model.mat detailing the nets will be stored.
-
func_FeatureSelection: This will find activation maps at the ReLu layer, perform Region Accumulation (RA) step and Element Decoder step to find gene subset. The input is model.mat (fromfunc_TrainModel) and related .mat file from the folder DeepResults. The net used is squeeznet. However, if different nets are used then netName at Line 6 of func_FeatureSelection.m file should be changed accordingly.
-
-
Non-image to image conversion: two core sub-functions of
func_Prepare_Dataare used to convert samples from non-image to image. These are described below.-
Cart2Pixel: The input to this function is the entire Train set. The output is the feature or gene locations Z in the pixel frame. The size of the pixel frame is pre-defined. -
ConvPixel: The input is a non-image sample or feature vector and Z (from above). The output is an image sample corresponding to the input sample.
-
-
Compression Snow-fall algorithm (SnowFall.m): The compression algorithm is used to provide more space for features in the given pixel frame. Since the conversion from Cartesian coordinates system to the pixel frame depends on the pixel resolution, it becomes difficult to fit all the features without overlapping each other. This algorithm tries to create more space such that the overlapping of feature or gene location can be minimized. The input is the locations of genes or features with the pixel size information. The output is the readjusted image. It is up to the user to use Snow-fall compression or not.
-
Extraction of Gene Names (optional): This option is useful for enrichment analysis. Two files for extraction of genes are GeneNames_Extract.m and GeneNames.m. The list of names of genes is stored in
~/DeepFeature_pkg/Datafolder.After running DeepFeature results will be stored in corresponding RunY and StageX folders (where X and Y are integers 1,2,3…). If it is required to find the gene IDs/names of the obtained subset for each cancer type, then execute
GeneNames_Extractfunction. Go to Line 4, and set theOut_Stagesvariable. Since Stage 2 has been saved inside Run1 after executing DeepInsight the first time, useOut_Stages = 2. Then go to Line 5 and defineFileRun. For example, it is set asFileRun = ‘Run1’.The gene list per class will be generated. Since here we used 10 cancer-types, so 10 files will be generated. In addition, one file with all genes listed will be generated (e.g. GeneList_UnCmprss.txt). The results will be stored in
~/Models/RunY/StageXas RunYStageX.tar.gz and a folder with the same results will also be created as RunYStageX. In this example, it will be stored in the folderRun1Stage2and Run1Stage2.tar.gz. -
Combining subsets of genes (optional): gene subsets can be combined, i.e., the union of individual gene lists obtained from different runs. In the paper, different gene subsets were combined to have a more comprehensive selection of genes for different distances used in tSNE. If a user wants to combine or have a union of genes/features then GenesFromRuns.m can be executed. Please select the gene lists by defining their path (e.g. at Line 5, line 19 if 2 gene subsets are to be combined). The overall combined gene list and combined lists for each cancer-type or class will be stored. The gene names will be stored following the TCGA file given. However, for your data, place the file in Data folder, and change Line 91 corresponding to the name of your file.
Parameter settings to run the package
A number of parameters/variables are used to control the DeepFeature_pkg. The details are given hereunder
-
Parm.Method(selection dimensionality reduction technique)Dimensionality reduction technique can be considered as one of the following methods; 1) tSNE 2) Principal component analysis (PCA) 3) kernel PCA, 4) uniform manifold approximation and projection (umap). For umap you can use python or R scripts (please see umapa_Rmatlab.m).
Select this variable in DeepFeature.m file (Line 4) as
Parm.Method = ‘tSNE’, ‘kpca’, ‘pca’ or ‘umap’
Default is tSNE.
-
Parm.Dist(Distance selection)If tSNE is used, then one of the following distances can be used. The default distance is ‘cosine’.
Parm.Dist = ‘cosine’, ‘hamming’, ‘mahalanobis’, ‘educidean’, ‘chebychev’, ‘correlation’, ‘minkowski’, ‘jaccard’, or ‘seuclidean’ (standardized Eucliden distance).
-
Parm.Max_Px_Size(maximum pixel frame either row or column)The default value is 227 as required by SqueezeNet architecture.
-
Parm.ValidRatio(ratio of validation data and training data)The amount of training data required to be used as a validation set. Default is 0.1; i.e., 10% of training data is kept aside as a validation set. The new training set will be 90% of the original size.
-
Parm.SeedRandom parameter seed to split the data.
-
Parm.FileRunChange the name as RunX, where X is an integer defining the run of DeepFeature on your data.
Change the value X for new runs.
-
Parm.SnowFall(compression algorithm)Suppose SnowFall compression algorithm is used then set the value as 1, otherwise 0. Default is set as 1.
-
Parm.Threshold(for Class Activation Maps)Set the threshold of class activation maps (CAMs) by changing the value between 0 and 1. If the value is high (towards 1), then the region of activation maps will be very fine. On the other hand, the region will be broader towards value 0. Default is 0.6. However, 0.45 was also used to produce some results in the paper.
-
Parm.DesiredGenesExpected number of genes to be selected. Default is set as 1200. However, change as required.
-
Parm.UsePrevModelDeepFeature is running in multiple stages. If you want to avoid running CNN multiple times then set these values as ‘y’ (yes); i.e., the previous weights of CNN will be used for the current model. This way, the processing time is shorter, however, performance (in terms of selection and accuracy) would be lower. The default setting is ‘n’ (no).
-
Parm.SaveModelsFor saving models type ‘y’, otherwise ‘n’. Default is set as yes ‘y’.
-
Parm.StageDefine the stage of execution. If you are running DeepFeature on your new data, then put
Parm.Stage=1. All the results will be saved in RunXStage1. If DeepFeature continues with the loop to find a smaller number of genes then Stage2, Stage3…, will be automatically processed and results will be saved in RunXStage2, RunXStage3,…, and so on.In the example, Stage2 is used because some genes are prefiltered and the row information of the selected ones was given in
Run1/Stage1/. -
Paths
Default paths for FIGS, Models and Data are
~/DeepFeature/FIGS/,~/DeepFeature/Models/and~/DeepFeature/Data/, respectively. Runtime parameters will be stored in~/DeepFeature/folder (such as model.mat, Out1.mat or Out2.mat). -
Log and performance file (including an overview of parameter information)
The runtime results will be stored in
~/DeepFeature/DeepFeature_Results.txtwith complete information about the run.
DeepInsight
A YouTube video about the original DeepInsight method is available here. A Matlab page on DeepInsight can be viewed from here.